💻 Usage¶
Inference usage¶
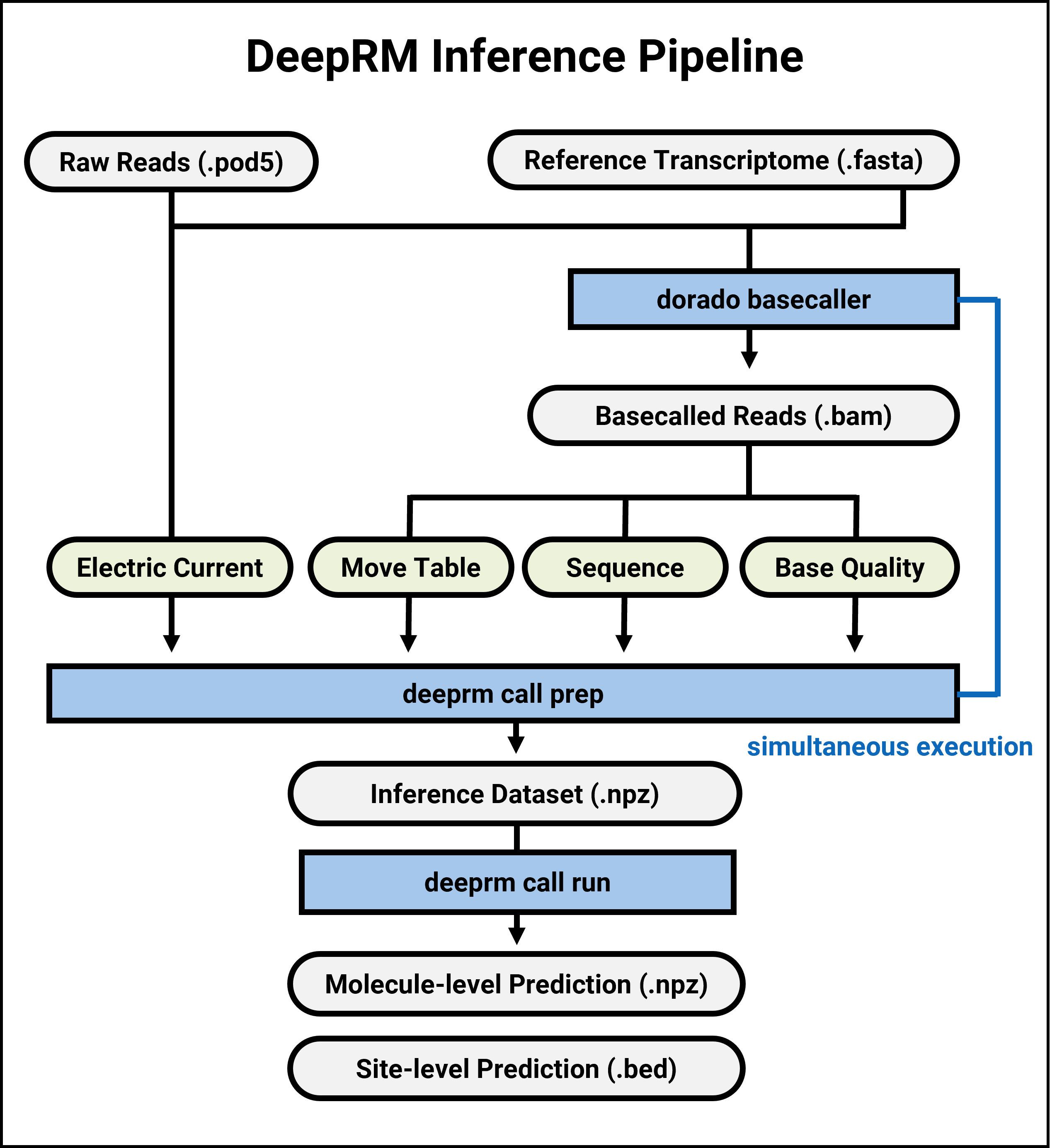
Prepare Data¶
Accelerated preparation (recommended, default)¶
This method uses precompiled C++ binary for accelerating the preprocessing step.
dorado basecaller --reference <ref_fasta> --min-qscore 0 --emit-moves rna004_130bps_sup@v5.0.0 <pod5_dir> \ | tee >(samtools sort -@ <threads> -O BAM -o <bam_path> - && samtools index -@ <threads> <bam_path>) \ | deeprm call prep -p <pod5_dir> -b - -o <prep_dir>
If Dorado fails due to “illegal memory access”, try adding
--chunksize <chunk_size>option (e.g., chunk_size=12000).If the precompiled binary does not work on your system, please refer to the advanced-installation page for detailed build instructions.
Adjust the
-g (--filter-flag)parameter according to your needs. If using a genomic reference, you may want to use-g 260.
Sequential preparation¶
This method is slower than the accelerated preparation method, but is supported for cases such as:
The POD5 files are already basecalled to BAM files with move tags.
You want to run basecalling and preprocessing in separate machines.
Basecall the POD5 files to BAM files with move tags (skip if already done):
If Dorado fails due to “illegal memory access”, try adding
--chunksize <chunk_size>option (e.g., chunk_size=12000).
dorado basecaller --reference <reference_path> --min-qscore 0 --emit-moves rna004_130bps_sup@v5.0.0 <pod5_dir> > <raw_bam_path>"
Filter, sort, and index the BAM files:
Adjust the
-Fparameter according to your needs. If using a genomic reference, you may want to use-F 260.
samtools view -@ <threads> -bh -F 276 -o <bam_path> <raw_bam_path>
samtools sort -@ <threads> -o <bam_path> <bam_path>
samtools index -@ <threads> <bam_path>
To preprocess the inference data (transcriptome), run the following command:
deeprm call prep -p <input_POD5_dir> -b <bam_path> -o <prep_dir>
This will create the npz files for inference.
Run Inference¶
The trained DeepRM model file is attached in the repository:
weight/deeprm_weights.pt.For inference, run the following command:
Modify the ‘-s’ (batch size) parameter according to your GPU memory capacity (default: 1000).
deeprm call run --model <model_file> --data <data_dir> --output <prediction_dir> --gpu_pool <gpu_pool>
This will create a directory with the result files.
Optionally, if you used a transcriptomic reference for alignment, you can convert the result to genomic coordinates by supplying a RefFlat/GenePred/RefGene file (
--annot <annotation_file>).
Site-level BED file format¶
The output BED file follows the standard bedMethyl format. Please see https://genome.ucsc.edu/goldenpath/help/bedMethyl.html for description.
Please note that columns 14 to 18 are zero-filled for compatibility. These columns will be used for a planned future update.
Molecule-level BAM file format¶
The output BAM file contains modification information in MM and ML tags. Please see https://samtools.github.io/hts-specs/SAMtags.pdf for description.
Molecule-level NPZ file format (advanced usage)¶
The output NPZ file contains the following arrays:
1. read_id
2. label_id
3. pred: modification score (between 0 and 1)
Read ID specification:
The UUID4 format read ID (128 bits) is converted to two 64-bit integers for NumPy compatibility.
You can convert the two 64-bit integers back to UUID4 using the following Python code:
import numpy as np import uuid def int_to_uuid(high, low): return uuid.UUID(bytes=b"".join([high.tobytes(),low.tobytes()]))
Label ID specification:
Label ID contains the reference, position, and strand information.
You can decode the label ID using the following Python code:
import numpy as np def decode_label_id(label_id, label_div = 10**9): strand = np.sign(label_id) label_id_abs = np.abs(label_id) - 1 ref_id = label_id_abs // label_div pos = label_id_abs % label_div return ref_id, pos, strand
Reference ID is extracted from the input BAM file header.
Training usage¶

Prepare Data¶
You can skip this step if your POD5 files are already basecalled to BAM files with move tags.
dorado basecaller --min-qscore 0 --emit-moves rna004_130bps_sup@v5.0.0 <pod5_dir> > <bam_path>
samtools index -@ <threads> <bam_path>
To preprocess the training data (synthetic oligonucleotide), run the following command:
deeprm train prep --input <input_POD5_dir> --output <output_file>
This will create:
Training dataset: /block
To compile the training dataset, run the following command:
deeprm train compile --input <input_POD5_dir> --output <output_file>
This will create:
Training dataset: /block
Run Training¶
To train the model, run the following command:
deeprm train run --model deeprm_model --data <data_dir> --output <output_dir> --gpu_pool <gpu_pool>
This will create a directory with the trained model file.